Nanoformulation Analytical Method Validation and Transfer
Inquiry
Validation of analytical methods for nanoformulations is crucial to ensure their reliability. The transfer of these methods involves moving them from laboratory settings to GMP-controlled environments for production. It is essential to maintain quality control of nanoformulations using dependable analytical methods throughout their life cycle, including development, production, transportation, and sales. CD Formulation places significant emphasis on researching and facilitating the verification and transfer of analytical methods for nanoformulations. We can support researchers in seamlessly integrating these processes into product development, production, and clinical transformation.
What is Nanoformulation Analytical Method Validation and Transfer?
Nanoformulation Analytical Method Validation
Nanoformulation analytical method validation is the establishment of documented evidence that provides a high degree of assurance that a specific process will consistently produce nanoformulations that meet their predetermined specifications and quality attributes.
Nanoformulation Analytical Method Transfer
Nanoformulation analytical method transfer is the documented process that qualifies a receiving laboratory to use a nanoformulation analytical method that originated in the transferring laboratory.
Our Nanoformulation Analytical Method Validation Process
CD Formulation has rich experience in exploring and researching nanoformulation analytical method validation and can provide our customers with our professional nanoformulation analytical method validation of main test items, such as particle size, encapsulation efficiency, drug loading, in vitro drug release, etc.
Our Nanoformulation Analytical Method Validation Indicators
To ensure that the analytical methods of the following test items are accurate and robust, we can perform the nanoformulation analytical method validation of the following test items according to different testing methods.
- Particle Size
- Encapsulation Efficiency
- Drug Loading
- Nanoparticle Concentration
- In Vitro Drug Release
- Elemental Impurity
- Residual Solvent
Nanoformulation Analytical Method Validation Characteristics
We perform nanoformulation analytical method validation according to ICH Q2. There are the following typical analytical method validation characteristics in ICH Q2.
Table 1 Typical performance characteristics and associated validation tests to measure product attributes. (ICH Q2)
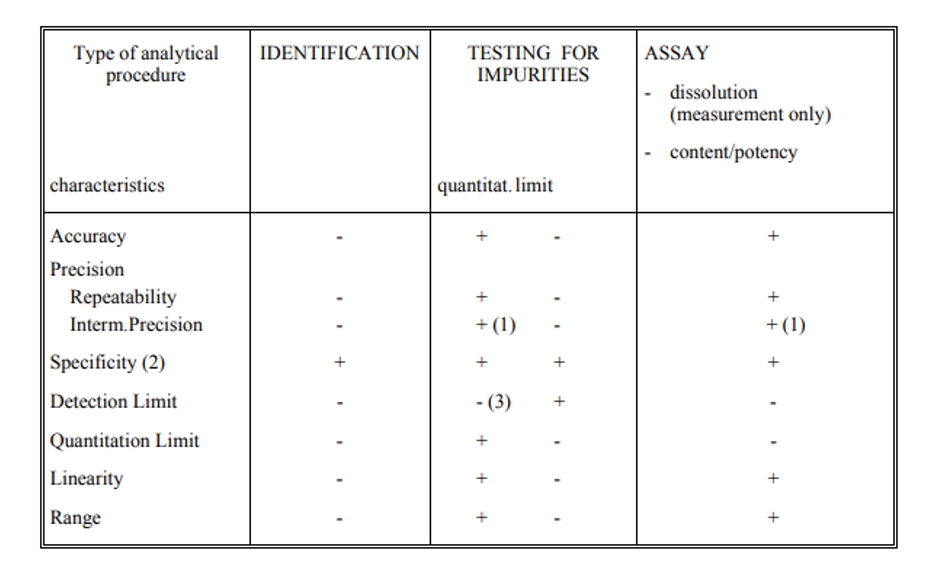
- signifies that this characteristic is not normally evaluated.
+ signifies that this characteristic is normally evaluated.
(1) in cases where reproducibility (see glossary) has been performed, intermediate precision is not needed.
(2) lack of specificity of one analytical procedure could be compensated by other supporting analytical procedure(s).
(3) may be needed in some cases.
Our Nanoformulation Analytical Method Transfer Process
The essence of nanoformulation analytical method transfer is to confirm, through the nanoformulation analytical method transfer program, that the receiving laboratory has the ability to develop methods and obtain the same accuracy and precision as the transferring laboratory.
Our Workflow of Nanoformulation Analytical Method Transfer
We can perform nanoformulation analytical method transfer through the following process.
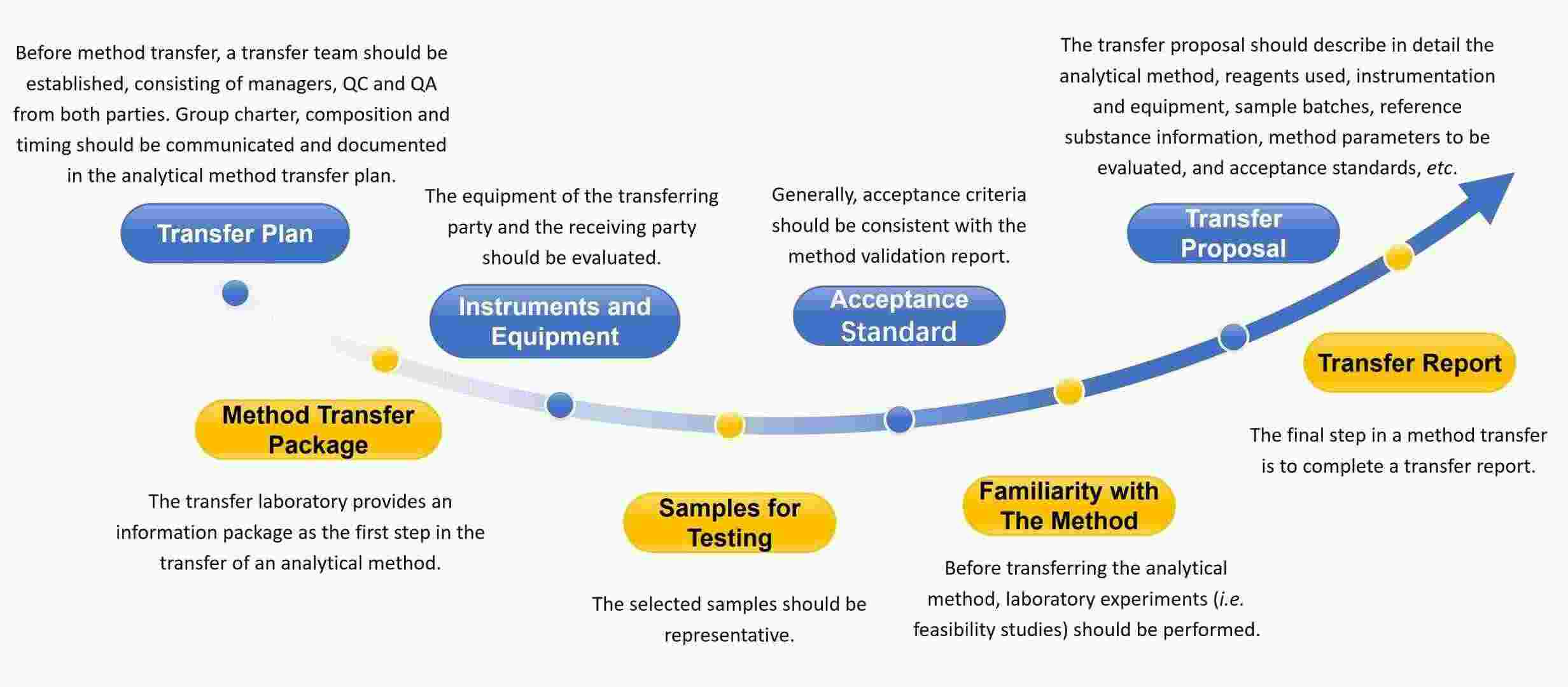 Fig.1 Our workflow of nanoformulation analytical method transfer. (CD Formulation)
Fig.1 Our workflow of nanoformulation analytical method transfer. (CD Formulation)
Types of Nanoformulation Analytical Method Transfer
There are four types of nanoformulation analytical method transfer as follows.
Comparative testing is to let the transferor and the recipient complete the testing of a batch of samples in a reasonably short time, and then compare and evaluate the results of each laboratory according to the acceptance criteria specified in the plan.
Covalidation between two or more laboratories: In most cases, the transferor laboratory takes precedence over the recipient laboratory in validating the method. This is a good option for transferring the nanoformulation analytical method from the company's R&D department to the QC laboratory.
Full validation of a method by the receiving laboratory is a common approach that allows the receiving laboratory to produce acceptable results. This approach is often used in the early stages or to correct or complete validation elements that were not previously performed as required by the regulations.
Transfer waiver: If this method is used, the reasons should be recorded and explained, such as the receiving laboratory has tested similar samples and is familiar with the test procedures. Analysts familiar with the method are transferred to the receiving laboratory, etc. Before the transfer of analytical methods, a risk assessment should also be conducted to determine the factors that may affect, rate the risks, and determine how to eliminate the risks.
Why Choose Us for Nanoformulation Analytical Method Validation and Transfer?
- Relying on our nanoformulation analytical technology platform, we can offer nanoformulation analytical validation and transfer to ensure the final analytical methods are accurate and robust.
- Our core analytical team has rich experience in nanoformulation analytical method development, validation and transfer, and we can quickly provide you with our professional proposal regarding your detailed requirements.
- We have complete analysis and testing service management specifications, a relatively fast sample circulation cycle, and can offer the nanoformulation analytical method validation of main test items, covering particle size, encapsulation efficiency, in vitro drug release, nanoformulation elemental impurities, residual solvents, etc.
Published Data
Technology: Analytical Method Development and Validation of RP-HPLC Method of Exemestane and Genistein
Journal: ACS Omega
IF: 4.1
Published: 2023
Results:
The authors quantified the precise concentrations of exemestane (EXE) and genistein (GEN) when provided as a combination to develop and validate an RP-HPLC method. Meanwhile, the Box-Behnken design was selected as the experimental model and the interaction between the independent and dependent variables was analyzed. Parameters such as linearity, system suitability, specificity, precision (intra-day and inter-day), robustness, durability, LOD (limit of detection), and LOQ (limit of quantification) were selected to validate the method proposed by the authors. The results showed that the method developed and validated by the authors was suitable for the simultaneous identification of the combination of these two drugs as well as the nanoformulation.
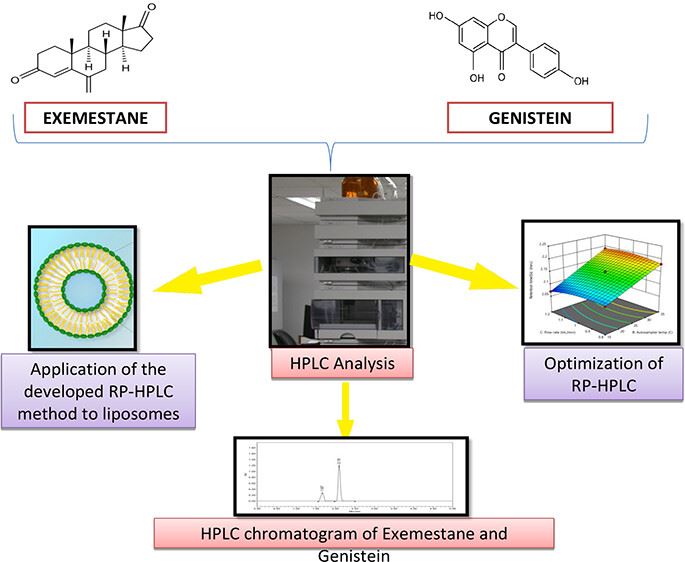 Fig.2 The analytical method development and validation process of Exemestane and Genistein. (Shwetakshi Sharma, et al. 2023)
Fig.2 The analytical method development and validation process of Exemestane and Genistein. (Shwetakshi Sharma, et al. 2023)
CD Formulation has accumulated rich experience in nanoformulation analytical method validation and transfer, and can quickly confirm the test methods, key risk factors, feasibility studies and on-site training according to the specific requirements of customers and guide the smooth completion of method transfer. If you are interested in our nanoformulation analytical method validation and transfer services, please kindly contact us for in-depth communication.
References
- ICH Q2(R2): Validation of Analytical Procedures.
- Shwetakshi Sharma, Priya Gupta, Annie Gupta, et al. Rapid Analytical Method Development and Validation of RP-HPLC Method for the Simultaneous Estimation of Exemestane and Genistein with Specific Application in Lipid-Based Nanoformulations. ACS Omega. 2023, 8, 25101-25113.
How It Works
STEP 2
We'll email you to provide your quote and confirm order details if applicable.
STEP 3
Execute the project with real-time communication, and deliver the final report promptly.
Related Services
